Auf TCT vorgestellte Studien bringen höhere Überlebensrate mit früher Erkennung von Rechtsherzinsuffizienz und frühem Einsatz von Impella RP in Verbindung
Auf TCT vorgestellte Studien bringen höhere Überlebensrate mit früher Erkennung von Rechtsherzinsuffizienz und frühem Einsatz von Impella RP in Verbindung
DANVERS, Massachusetts (USA)--(BUSINESS WIRE)--Auf der TCT Connect, dem 32. jährlichen wissenschaftlichen Symposium der Cardiovascular Research Foundation, vorgestellte Daten zeigen, dass eine frühzeite Erkennung der Rechtsherzinsuffizienz und die frühzeitige Anwendung von Impella RP mit signifikant höheren Überlebensraten einhergehen. Die frühe Erkennung von Patienten, die eine Unterstützung des rechten Herzens benötigen, ist von entscheidender Bedeutung, da frühere Studien gezeigt haben, dass 37 % der Patienten mit Myokardinfarkt-bedingtem kardiogenem Schock (AMICS) eine Rechtsherzinsuffizienz1 aufweisen, was zu einem achtmal höheren Sterblichkeitsrisiko führt2.
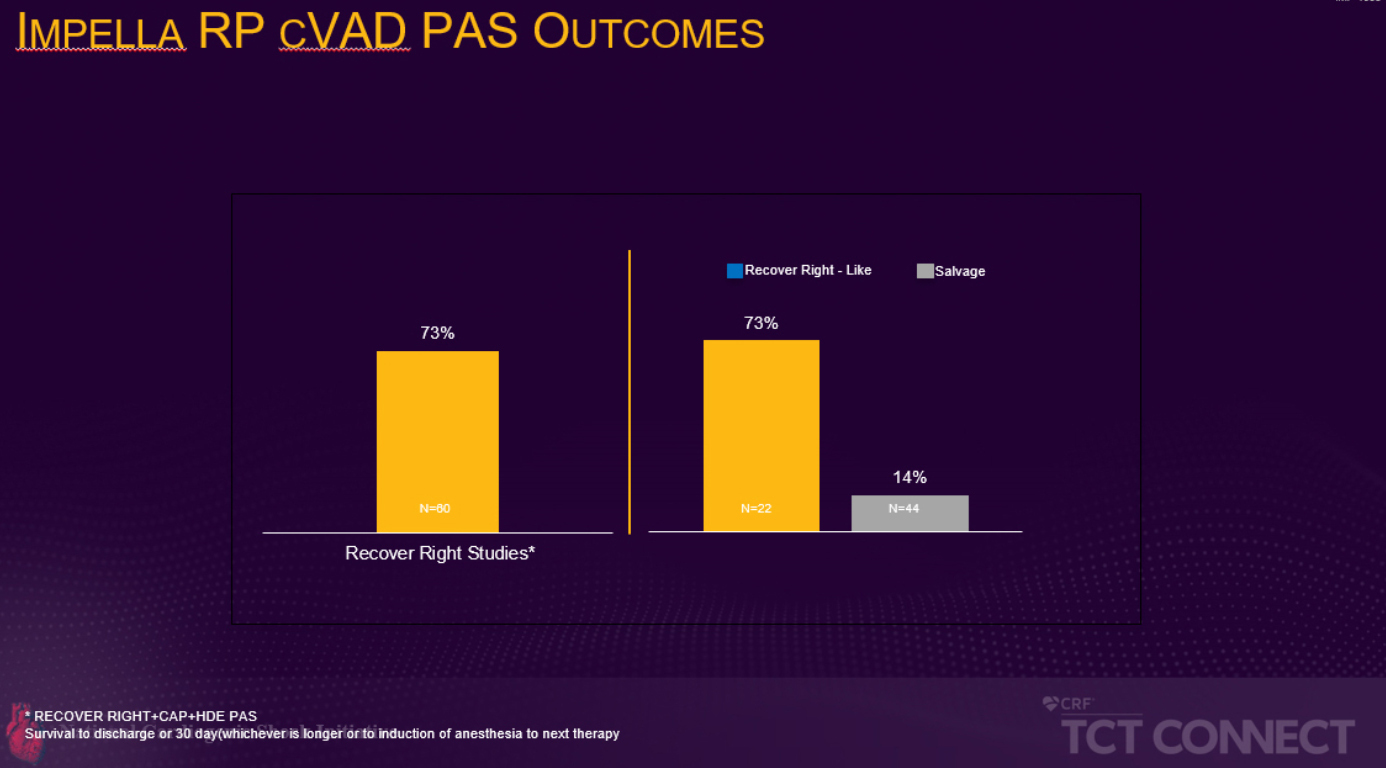
Die prospektive, multizentrische FDA-PMA-Post-Approval-Studie, die im Rahmen der TCT vorgestellt wurde, verglich die Überlebensrate von Patienten, die die Aufnahmekriterien für die RECOVER RIGHT-Studie erfüllt hätten, mit der von Patienten, die sich nicht für die Studie qualifiziert hätten, weil sie sich mehr als 48 Stunden im Zustand des kardiogenen Schocks befanden. Die Daten der RECOVER RIGHT-Studie und der anschließenden HDE-Post-Approval-Studie wurden zwischen 2012 und 2017 gesammelt und führten im Jahr 2017 zu einer PMA-Zulassung für die Impella RP. Die laufende PMA-Post-Approval-Studie, die auf der TCT Connect vorgestellt wurde, rekrutierte Patienten von September 2017 bis Juni 2019 und ergab, dass Patienten, die innerhalb von 48 Stunden nach Beginn des kardiogenen Schocks eine Impella RP-Unterstützung erhielten, hatten eine deutlich höhere Überlebensrate als Patienten, die eine verzögerte Rechtsherzunterstützung erhielten (73 % vs. 14 %, p<0,001). Die 73% Überlebensrate ist vergleichbar mit der Überlebensrate in den der PMA vorausgehenden Studien RECOVER RIGHT und HDE (siehe Abbildung 1).
„Die frühzeitige Erkennung der Rechtsherzinsuffizienz und frühzeitige Maßnahmen sind der Schlüssel zur Verbesserung der Überlebensraten der Patienten“, sagte Dr. Mark Anderson, Vorsitzender der Abteilung für Herzchirurgie am HUMC/Hackensack Meridian Health. „Diese Studie deutet darauf hin, dass bei Rechtsherzinsuffizienz die Verkürzung der Zeit zwischen Schockbeginn und Einführung der Impella RP ein Schlüsselelement für eine angemessene Patientenunterstützung ist".
Eine zweite Studie, die im Rahmen der TCT vorgestellt wurde, soll Kliniker dabei unterstützen, frühe Auslöser der Rechtsherzinsuffizienz zu erkennen. Die Analyse von 100 Patienten, die von den leitenden Prüfärzten der National Cardiogenic Shock Initiative (NCSI)-Studie durchgeführt wurde, verglich AMICS-Patienten mit rechtsventrikulärer Insuffizienz (RVF) mit Patienten ohne und stellte fest, dass anhaltende diastolische Saugalarme auf dem Automated Impella Controller (AIC) und ein erhöhter zentraler Venendruck (CVP) von mehr als 12 mmHg ein früher Hinweis auf eine RVF sein können (siehe Abbildung 2).
„Der Echtzeit-Einblick des AIC kann ein wichtiges Instrument sein, um den Arzt dabei zu unterstützen, bessere Behandlungsergebnisse für die Patienten zu erzielen“, sagte Babar Basir, DO, interventioneller Kardiologe am Henry Ford Hospital. „Länger anhaltende diastolische Saugalarme können bei Patienten mit erhöhtem Füllungsdruck ein frühes Anzeichen für eine rechtsventrikuläre Insuffizienz sein, und eine längere Dauer des diastolischen Sogs ist mit schlechteren Ergebnissen verbunden.“
Impella RP ist das am besten untersuchte Gerät für die rechte Seite und die einzige perkutane Technologie mit einer FDA-Zulassung, die es als sicher und wirksam für die Unterstützung des rechten Herzens einstuft. Die exklusive FDA-Zulassung ist das Ergebnis von fünf Jahren Forschung, die Folgendes umfasste:
- RECOVER RIGHT, eine von der FDA genehmigte, prospektive, multizentrische, einarmige, multizentrische Studie, die begann, nachdem das Unternehmen im November 2012 die Genehmigung der FDA für die Freistellung von Prüfpräparaten (Investigational Device Exemption, IDE) erhalten hatte, und 2014 abgeschlossen wurde.
- Ein Protokoll für kontinuierlichen Zugang (Continuous Access Protocol, CAP)
- HDE-Post-Approval-Studie, 2017 abgeschlossen
- PMA-Post-Approval-Studie, Beginn im September 2017
Darüber hinaus erteilte die FDA am 29. Mai 2020 eine Notfallzulassung (Emergency Use Authorization, EUA), um den Einsatz von Impella RP auf Patienten auszuweiten, die an COVID-19-bezogenen rechtsventrikulären Komplikationen leiden, einschließlich rechtsventrikulärer Dysfunktion im Zusammenhang mit einer Lungenembolie. Impella ist das einzige kardiovaskuläre Therapiegerät, das von der FDA die Notfallzulassung für die Behandlung von COVID-19-Patienten erhalten hat.
________________
1 Lala et al. J Card Fail. 2018;24:148-156
2 Mehta et al. J Am Coll Cardiol. 2001;37:37-43
ÜBER IMPELLA HERZPUMPEN
Die Geräte Impella 2.5® und Impella CP® haben von der US-Gesundheitsbehörde FDA eine PMA-Zulassung zur Behandlung bestimmter Patienten mit fortgeschrittener Herzinsuffizienz erhalten, bei denen elektive und dringende perkutane Koronarinterventionen (PCI) wie Stenting oder Ballonangioplastie zur Öffnung blockierter Koronararterien durchgeführt werden. Bei den Modellen Impella 2.5, Impella CP, Impella CP mit SmartAssist®, Impella 5.0®, Impella LD® und Impella 5.5® mit SmartAssist® handelt es sich um FDA-zugelassene Herzpumpen, die zur Behandlung von Herzinfarkt- oder Kardiomyopathie-Patienten im kardiogenen Schock eingesetzt werden und über die einzigartige Fähigkeit verfügen, eine native Wiederherstellung des Herzens zu ermöglichen, so dass die Patienten mit ihrem eigenen Herzen nach Hause entlassen werden können. Die Impella RP® ist in den USA von der FDA zur Behandlung von Rechtsherzinsuffizienz oder Dekompensation nach Implantation eines Linksherzunterstützungssystems, Myokardinfarkt, Herztransplantation oder Operation am offenen Herzen zugelassen. Das Impella RP® ist auch für den Notfalleinsatz im Krankenhaus für die vorübergehende rechtsventrikuläre Unterstützung für bis zu 14 Tage bei Intensivpatienten mit einer Körperoberfläche von ≥1,5 m2 zur Behandlung von akuter Rechtsherzinsuffizienz oder Dekompensation aufgrund von Komplikationen im Zusammenhang mit der Coronavirus-Krankheit 2019 (COVID-19), einschliesslich Lungenembolie (PE), zugelassen. Impella RP ist nicht für die Behandlung der akuten Rechtsherzinsuffizienz oder Dekompensation aufgrund von Komplikationen im Zusammenhang mit COVID-19 freigegeben oder zugelassen. Die Impella Linksherzunterstützungssysteme (LV-Unterstützungssysteme) sind auch für den Notfalleinsatz durch Gesundheitsdienstleister in Krankenhausumgebungen zur vorübergehenden Versorgung zugelassen (≤ 4 Tage für Impella 2.5, Impella CP und Impella CP mit SmartAssist; und ≤ 14 Tage für Impella 5.0 und Impella 5. 5 mit SmartAssist) zur LV-Entladung und Unterstützung zur Behandlung von Intensivpatienten mit bestätigter COVID-19-Infektion, die einer ECMO-Behandlung unterzogen werden und die während der V-A-ECMO-Unterstützung ein Lungenödem oder während der V-V-ECMO-Unterstützung eine späte Herzdekompensation durch Myokarditis entwickeln. Die autorisierten Impella LV-Unterstützungssysteme sind für die zugelassene Indikation weder freigegeben noch für den Einsatz zugelassen. Die Impella RP- und Impella LV-Unterstützungssysteme wurden von der FDA im Rahmen einer EUA für die oben genannte Notfallverwendung genehmigt und nur für die Dauer der Erklärung, dass Umstände vorliegen, die die Genehmigung der Notfallverwendung von Medizinprodukten gemäß Abschnitt 564(b)(1) des Gesetzes, 21 U.S.C. § 360bbb-3(b)(1) rechtfertigen, es sei denn, die Genehmigung wird früher aufgehoben oder widerrufen.
In Europa sind Impella 2.5, Impella CP und Impella CP mit SmartAssist CE-gekennzeichnet für die Behandlung von Hochrisiko-PCI- und AMI-Patienten mit kardiogenem Schock bis zu 5 Tagen. Impella 5.0 und Impella LD sind CE-gekennzeichnet für die Behandlung von Patienten mit Herzinfarkt oder Kardiomyopathie im kardiogenen Schock für bis zu 10 Tage. Impella 5.5 mit SmartAssist ist CE-gekennzeichnet, um Herzinfarkt- oder Kardiomyopathie-Patienten im kardiogenen Schock bis zu 30 Tage lang zu behandeln. Impella RP ist CE-gekennzeichnet zur Behandlung von Rechtsherzinsuffizienz oder Dekompensation nach Implantation eines Linksherzunterstützungssystems, Myokardinfarkt, Herztransplantation, Operation am offenen Herzen oder refraktärer ventrikulärer Arrhythmie. Weitere Informationen über Impella Herzpumpen, einschließlich der zugelassenen Indikationen und wichtiger Sicherheits- und Risikoinformationen im Zusammenhang mit der Verwendung der Geräte, finden Sie unter www.impella.com.
ÜBER ABIOMED
Abiomed, Inc. mit Sitz in Danvers, Massachusetts, USA, ist ein führender Anbieter von Medizinprodukten zur Kreislaufunterstützung. Unsere Produkte sind so konzipiert, dass das Herz zur Ruhe kommt, indem sie den Blutfluss verbessern und/oder die Pumpleistung des Herzens unterstützen. Weitere Informationen finden Sie unter www.abiomed.com. Abiomed, Impella, Impella 2.5, Impella 5.0, Impella 5.5, Impella LD, Impella CP, Impella RP, SmartAssist und Impella Connect sind eingetragene Warenzeichen von Abiomed, Inc. und sind in den USA und bestimmten anderen Ländern registriert. Impella BTR, Impella ECP, CVAD Study und STEMI DTU Study sind anhängige Warenzeichen von Abiomed, Inc.
ZUKUNFTSGERICHTETE AUSSAGEN
Diese Pressemitteilung enthält zukunftsgerichtete Aussagen, einschließlich Aussagen zur Entwicklung von bestehenden und neuen Produkten von Abiomed, zu den Fortschritten des Unternehmens im Bereich des kommerziellen Wachstums sowie zu zukünftigen Möglichkeiten und erwarteten behördlichen Zulassungen. Die tatsächlichen Ergebnisse des Unternehmens können erheblich von den in diesen zukunftsgerichteten Aussagen erwarteten Ergebnissen abweichen, was auf einer Reihe von Faktoren beruht, einschließlich Unsicherheiten im Zusammenhang mit Umfang, Ausmaß und Dauer der Auswirkungen der COVID-19-Pandemie, Entwicklungsaktivitäten, Tests und damit verbundenen behördlichen Zulassungen, einschließlich des Potenzials für zukünftige Verluste, komplexer Herstellung und hoher Qualitätsanforderungen, Abhängigkeit von begrenzten Bezugsquellen, Wettbewerb, technologischer Wandel, staatliche Regulierung, Rechtsstreitigkeiten, zukünftiger Kapitalbedarf und die Ungewissheit einer zusätzlichen Finanzierung sowie andere Risiken und Herausforderungen, die in den Einreichungen des Unternehmens bei der Securities and Exchange Commission (SEC) im Einzelnen aufgeführt sind, einschließlich des zuletzt eingereichten Jahresberichts auf Formular 10-K und der später bei der SEC eingereichten oder bei ihr eingereichten Unterlagen. Die Leser werden davor gewarnt, sich in unangemessener Weise auf zukunftsgerichtete Aussagen zu verlassen, die nur zum Zeitpunkt dieser Pressemitteilung Gültigkeit haben. Das Unternehmen verpflichtet sich nicht, die Ergebnisse von Berichtigungen dieser zukunftsgerichteten Aussagen zu veröffentlichen, die vorgenommen werden, um Ereignisse oder Umstände widerzuspiegeln, die nach dem Datum dieser Pressemitteilung eintreten, oder um das Eintreten unvorhergesehener Ereignisse widerzuspiegeln.
Die Ausgangssprache, in der der Originaltext veröffentlicht wird, ist die offizielle und autorisierte Version. Übersetzungen werden zur besseren Verständigung mitgeliefert. Nur die Sprachversion, die im Original veröffentlicht wurde, ist rechtsgültig. Gleichen Sie deshalb Übersetzungen mit der originalen Sprachversion der Veröffentlichung ab.
Contacts
Tom Langford
Director of Communication
Tel.: (978) 882-8408
TLangford@abiomed.com